Interactive Essential Dynamics (IED) provides fully interactive visualization of essential dynamics (ED) results within Visual Molecular Dynamics (VMD). It reads output from ED analysis performed in GROMACS or Ptraj. It is also possible to perform an ED calculation directly with IED. IED was programmed by John Mongan <jmongan |at| mccammon |dot| ucsd |dot| edu >, and is free for download and use under the GNU Lesser General Public License (LGPL). Comments from users are welcome (please include IED in the subject of your email).
Improvements in this version include:
Download IED here.
You may also want to download sample
essential dynamics results files.
In addition, you will need to download and install any of the following that you don't already have. Note that you must install exactly the same versions of Python and Numeric that your VMD was compiled against, and Numeric must be installed into whichever Python install VMD is using (there may be more than one on your system).
If you are using a VMD version downloaded from the VMD website rather than compiling your own version from source, the easiest way to get compatible Python and Numeric is from the VMD python library page.
When you have successfully installed all of the above, you should
be able to start VMD and type the following into the console window
gopython import vmdnumpy,Numericwithout any error messages appearing. If you get error messages, you do not have all the needed software properly installed and configured, and IED is unlikely to work.
It is strongly recommended that you have at least one of the
following packages for performing ED analysis
IED has been tested with VMD 1.8.1 and 1.8.2, Python 2.2.2, NumPy 22, GROMACS 3.1 and ptraj 7.0 on Linux. More recent versions of these programs should also work. Note that VMD 1.8.2 has a bug which requires a work-around detailed below. This bug is not in 1.8.1 and should be fixed in the next release after 1.8.2.
If you find IED to be useful in conducting work that leads to publication, please cite it in your manuscript. IED is described in:
Interactive Essential Dynamics. John Mongan. Journal of Computer-Aided Molcular Design 2004. 18(6): 433-36
There are three options for performing the ED calculations: using
GROMACS, using ptraj or within IED itself. In general, GROMACS is
preferred for working with GROMACS format trajectories, and ptraj is
preferred for working with AMBER or CHARMM format trajectories.
Performing the calculations within IED is the easiest option, but the
matrix diagonalization step is significantly slower than with GROMACS
or ptraj, there is less flexibility in how the calculation is performed
and the results cannot be easily saved. As such, ED calculations within
IED may be most useful for small systems that can be calculated
quickly, for trajectory formats that cannot be read by ptraj or
GROMACS,
or for one-off calculations where installation of additional software
is undesirable.
When using GROMACS to prepare input for IED, one uses g_covar to find the eigenvectors and then g_anaeig to generate the projections of the trajectory onto the eigenvectors. For instance
g_covar -f <traj file> -s <topol file> -o eigenval.xmgr -v eigenvec.trr -av average.pdb
g_anaeig -v eigenvec.trr -f <traj file> -proj proj.xvg -last 10
Where <traj file>
is your molecular dynamics trajectory and <topol file> is a
topology file that corresponds to your trajectory. Note that the
eigen
decomposition performed by g_covar
can take a long time, and scales non-linearly with the number of
atoms. Also, the -last value
(10, in this case) given to
g_anaeig specifies how many eigenvectors will be available
through the IED interface.
The three files you will need are the eigenvector file (e.g.
eigenvec.trr), the
projections file (e.g. proj.xvg)
and a topology
file. If you performed essential dynamics using all atoms, your
topology file can be the same file passed to g_covar; if you used a
subset of the atoms, you need a topology file that matches the subset.
You can use g_anaeig to
generate the subset topology file like this
g_anaeig -v eigenvec.trr -f <traj file> -last 10 -filt subset-topol.pdb -b 1 -e 2
selecting the appropriate subset of atoms when prompted. Don't worry
about the coordinates that get written into the pdb file -- they're
ignored by IED.
If you are using VMD 1.8.2, you need to add a box to the eigenvec.trr file. Version
1.8.2 will not read a TRR format file with a zero box size. Earlier and
later versions of VMD do not seem to have this bug. You can add a box
using trjconv like this
trjconv -f eigenvec.trr -o eigenvecbox.trr -box 1 1 1
When using ptraj to prepare input for IED, one runs ptraj to find
the eigenvectors and then a second time to generate the projections.
(Hopefully it may be possible to combine this into a single run in
future versions of ptraj). Please note that only versions of ptraj
released prior to March 2004 do not include the features necessary for
ED calculations.
Ptraj is run by typing
ptraj <topology file>
at the command line, where <topology
file> is an AMBER prmtop file or a CHARMM psf file. Then a
script of commands is entered. Here is a script to generate the first
25 eigenvectors, where <trajectory
file> is an AMBER or CHARMM format trajectory:
trajin <trajectory file>
rms first *
matrix covar name mcovar
analyze matrix mcovar out evec.pev vecs 25
go
and here is a script to generate a corresponding set of projections:
trajin <trajectory file>
rms first *
projection modes evec.pev out proj.ppj beg 1 end 25
go
This produces the eigenvector file and projection file that you will
need to load into IED. Note that you can replace evec and proj with
whatever filenames you choose, but the extensions (.pev and .ppj) are
used to determine the file type and should not be changed. You will
also need a topology file for IED, which due to current limitations in
VMD, must contain coordinates (e.g. a PDB file works, but a prmtop file
does not). You can generate this PDB file using ptraj:
trajin <trajectory file> 1 1
trajout topol.pdb pdb
go
Start VMD and load IED (see Starting IED).
Load your trajectory. If you have not already "fit" your trajectory
(removed rotational and translational motion), you should do so now, or
you will see rotational and translational modes in the results of your
ED analysis. You can "fit" your trajectory by making sure it is the
"top" trajectory (it has a "T" next to it in the VMD main window) and
typing
source <path to IED>/vmdtrajal
trajal
into the VMD console. The first line will load a small Tcl script
included with IED and the second will perform the fit, printing the
RMSD for each frame as it goes. (Unfortunately, VMD currently does not
allow any way for IED to invoke this automatically; hopefully this may
change in future versions.)
From the IED main window, select the "Calculate ED from trajectory"
button. Enter the ID number of the trajectory you wish to analyze (ID
numbers are found on the left side of the VMD main window) and the
number of eigenvectors to display. Press "Calculate". As you will be
warned, VMD will freeze until the ED calculation completes. This can be
a long time, and scales nonlinearly with the number of atoms in the
system. There is no way to save the results of an ED calculation
performed within IED. If you anticipate needing to return to the
results of an analysis at a later date, it is better to use ptraj or
GROMACS.
Start VMD as you normally would (usually by typing vmd at a command prompt). When
vmd has loaded, type
gopython <path to IED>/ied.py
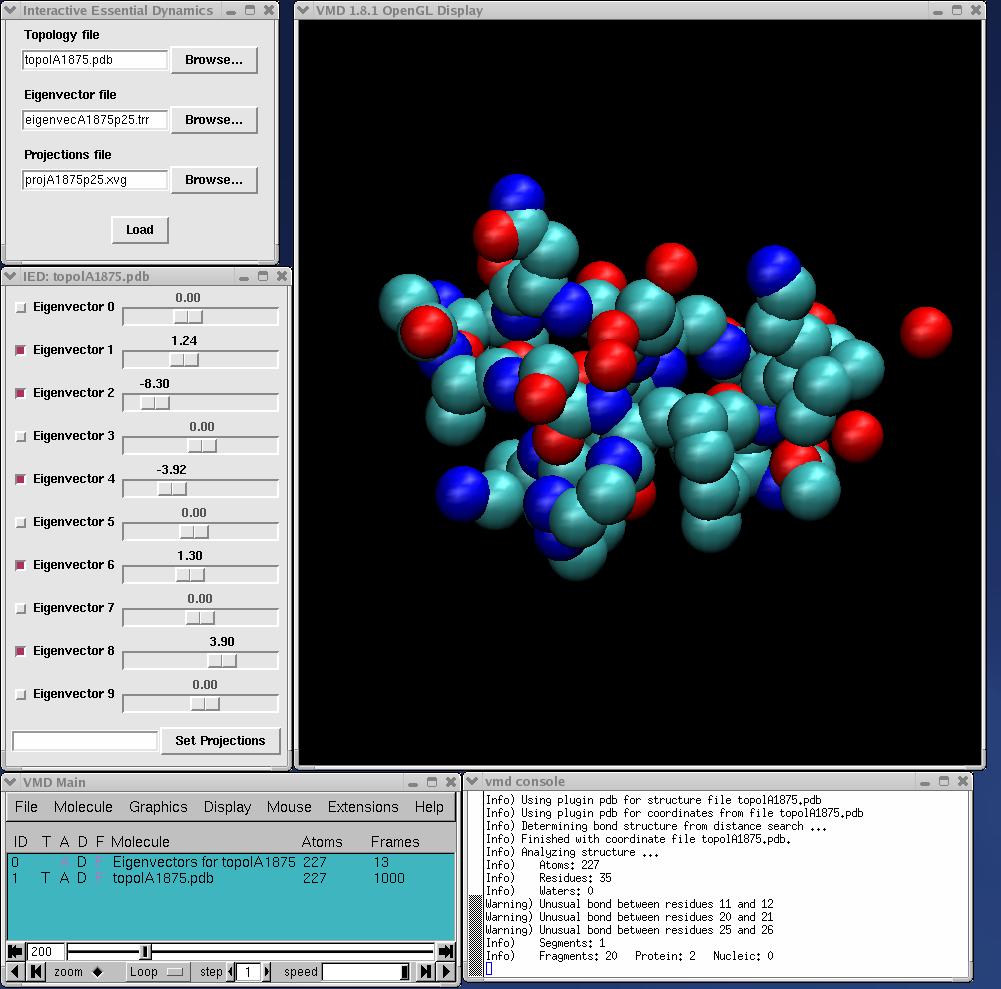
into the VMD console window (lower right window in the screen shot above), where <path to IED> is the location at which you have installed ied.py, e.g. /usr/local/bin/ied.py if it is in /usr/local/bin. If everything has been successfully installed, you should see the IED copyright notice in the VMD console and the main window, consisting of two buttons, will appear. If you have ED data from GROMACS or ptraj, press "Load ED data from files", and the file loader dialog will appear. Use this to select the appropriate topology, eigenvector and projections files. When you have located these files, click the load button. Note that file extensions are used to determine file type, so they must match the examples shown above. You may omit the projections file, in which case IED will allow manipulation of the structure along each vector, but will not enable the trajectory filtering feature. This may be of use when visualizing vectors derived from normal modes calculations, where there is no accompanying trajectory.
IED works in conjunction with VMD. All the standard features of VMD
remain available while using IED.
When the trajectory initially loads, all eigenvectors are deselected, and the molecule's conformation represents the average coordinates over the trajectory. Playing the trajectory will show no motion. Clicking a checkbox will turn on (or off) the corresponding eigenvector, allowing displacements along that eigenvector to be displayed. Eigenvectors can be turned on or off in any combination at any time. If you toggle an eigenvector while the trajectory is playing, there will be a short pause while coordinates are recalculated, after which the animation will resume. While the trajectory plays, sliders for the active eigenvectors will be moved to indicate each frame's projection on each active eigenvector.
When the trajectory is stopped, you can use the mouse to move the slider for any of the active eigenvectors. As the slider is moved, the molecular display is updated, illustrating the effect of changing the projection along the corresponding eigenvector. It is also possible to set the projection along all eigenvectors simultaneously by turning them all on, typing a comma delimited list of projections in to the text field and clicking the set projections button. Changes made to projection values by either of these methods are temporary: as soon as the frame is changed, they are lost. You can restore the original (trajectory) values for the projections by moving to a different frame and then returning to the original frame.
When exactly one eigenvector is selected (enabled), you can turn on the eigenvector drawing mode by clicking on the checkbox labled "Draw eigenvector on atom click". In this mode, when you click on an atom in the 3D display, an arrow will be drawn through it showing the direction of the atom's motion along the selected eigenvector. The point of the arrow represents the atom's position at the trajectory's maximum (most positive) projection and the tail represents the position at the minimum (most negative) projection. Note that VMD only registers atom clicks when you are in an atom selecting mouse mode (when the cursor is a crosshair in the display window). Typing 1 on the keyboard with the mouse cursor in the display window will put you into an appropriate mode. Selecting or deselecting any eigenvector will exit eigenvector drawing mode and erase the arrows.
These notes are of mostly historical interest, now that IED is able to read output from Ptraj. In general it will be easier to analyze AMBER trajectories with Ptraj than with GROMACS.
GROMACS can be used to analyze trajectories produced by the SANDER module of AMBER. GROMACS will read the AMBER trajectory if it is renamed to have an extension of .g87 . GROMACS will not read the AMBER prmtop topology file, but you can produce a pdb file to use as the topology file like this
ambpdb -p prmtop <prmcrd >topol.pdb
There is one problem with using GROMACS to analyze AMBER data: GROMACS assumes coordinates are in nanometers, while AMBER writes them in Angstroms.You can ignore this problem, in which case all your atoms will be ten times further apart than they should be in IED. Since VMD uses distances to determine bonding for PDB files, this means that if you use a PDB topology file, you won't see any bonds. If you use the van der Waals representation (with an increased radius) and remember to divide any distances you measure by 10, this may be acceptable.
If you are comfortable with recompiling GROMACS, you can use the following patch so that your distances will be correct and bonds will appear. Note that with this patch, all .g87 files are assumed to be in units of Angstroms, so if you think you may actually need to read a .g87 file in units of nanometers, this may not be a good idea.
The following patch should be applied to src/gmxlib/trxio.c
--- trxio.c 2002-02-28 02:49:31.000000000 -0800
+++ trxio.c 2003-12-12 17:11:34.000000000 -0800
@@ -453,14 +453,14 @@
/* else eof! */
return FALSE;
}
- x[i][m]=x0;
+ x[i][m]=x0 * 0.1;
}
}
if (bReadBox) {
for(m=0; (m<DIM); m++) {
if (fscanf(status,"%lf",&x0) != 1)
return FALSE;
- box[m][m]=x0;
+ box[m][m]=x0 * 0.1;
}
}
return TRUE;