|
 |
 | We have recently changed our website. Looking for the previous site? |



Abstracts of Articles in 2012
To request any journal reprints, please contact us.- Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition
- Adaptive Finite Element Modeling Techniques for the Poisson-Boltzmann Equation
- Introduction to Molecular Dynamics: Theory and Applications in Biomolecular Modeling
- Independent-Trajectory Thermodynamic Integration: A Practical Guide to Protein-Drug Binding Free Energy Calculations Using Distributed Computing
- Guide to Virtual Screening: Application to the Akt Phosphatase PHLPP
- A Molecular Dynamics Ensemble-Based Approach for the Mapping of Druggable Binding Sites
- Accelerated Molecular Dynamics in Computational Drug Design
- On the Use of Molecular Dynamics Receptor Conformations for Virtual Screening
- Hydrophobic Association and Volume-confined Water Molecules
- Mathematical and Numerical Aspects of the Adaptive Fast Multipole Poisson-Boltzmann Solver
- Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics Simulations
- Computer-aided Drug Discovery: Two Antiviral Drugs for HIV/AIDS
- Level-Set Variational Implicit-Solvent Modeling of Biomolecules with the Coulomb-Field Approximation
- AutoClickChem: Click Chemistry in Silico
- Accelerated Molecular Dynamics: Theory, Implementation and Application
- Simulations of the p97 complex suggest novel conformational states of hydrolysis intermediates
- Multiscale Continuum Modeling and Simulation of Biological Processes: From Molecular Electro-diffusion to Sub-Cellular Signaling Transduction
- Thermodynamic integration to predict host-guest binding affinities
- Dynamics and calcium association to the N-terminal regulatory domain of human cardiac Troponin C: A multi-scale computational study
- Comparative Molecular Dynamics Simulations of the Antimicrobial Peptide CM15 in Model Lipid Bilayers
- Modeling Cardiac Calcium Sparks in a Three-Dimensional Reconstruction of a Calcium Release Unit
- Nucleotide dependent mechanism of Get3 as elucidated from free energy calculations
- The Molecular Dynamics of Trypanosoma brucei UDP-Galactose-4’-Epimerase: A Drug Target for African Sleeping Sickness
- Hybrid Finite Element and Brownian Dynamics Method for 1-D Linear and Radial Diffusion-controlled Reactions
- Multi-timescale conformational dynamics of the SH3 domain of CD2-associated protein using NMR spectroscopy and accelerated molecular dynamics
- Novel cruzain inhibitors for the treatment of Chagas’s disease
- LigMerge: A Fast Algorithm to Generate Models of Novel Potential Ligands from Sets of Known Binders
- Structure Based Discovery of Novel Druggable Pockets on Rho Family GTPases
- Latest Advances in Classical Molecular Dynamics
- The dynamic structure of thrombin in solution
- iAPBS: A programming interface to Adaptive Poisson-Boltzmann Solver (APBS)
- Exploring the Photophysical Properties of Molecular Systems using Excited State Accelerated Ab Initio Molecular Dynamics
- Modeling effects of L-type Ca2+ current and Na+-Ca2+ exchanger on Ca2+ trigger flux in rabbit myocytes with realistic t-tubule geometry
- Spectroscopic and Computational Study of Melittin, Cecropin A, and the Hybrid Peptide CM15
- Calcium binding and allosteric signaling mechanisms for sarcoplasmic reticulum Ca2+ ATPase
- Routine access to millisecond timescale events with accelerated molecular dynamics
- Long timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in Troponin C
- Comment on “Molecular driving forces of the pocket-ligand hydrophobic association” by G. Graziano, Chem. Phys. Lett. 533 (2012) 95
- Dynamics of Plasmodium falciparum Enoyl-ACP Reductase and Implications on Drug Discovery
- The Binding Mechanism, Multiple Binding Modes, and Allosteric Regulation of Staphylococcus aureus SrtA Probed by Molecular Dynamics Simulations
- Finite Element Estimation of Protein-Ligand Association Rates with Post-Encounter Effects: Applications to Calcium binding in Troponin C and SERCA
- Molecular Basis of Calcium-sensitizing Mutations of the Human Cardiac Troponin C Regulatory Domain: A multi-scale simulation study
- Allosteric Networks in Thrombin Distinguish Procoagulant vs. Anticoagulant Activities
Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognitionQuarterly Reviews of Biophysics, Vol. 45, Issue 1, pp. 1-25 (2012) [PubMed 22082669]
Molecular recognition plays a central role in biochemical processes. Although well studied, understanding the mechanisms of recognition is inherently difficult due to the range of potential interactions, the molecular rearrangement associated with binding, and the time and length scales involved. Computational methods have the potential for not only complementing experiments that have been performed, but also in guiding future ones through their predictive abilities. In this review, we discuss how molecular dynamics (MD) simulations may be used in advancing our understanding of the thermodynamics that drive biomolecular recognition. We begin with a brief review of the statistical mechanics that form a basis for these methods. This is followed by a description of some of the most commonly used methods: thermodynamic pathways employing alchemical transformations and potential of mean force calculations, along with end-point calculations for free energy differences, and harmonic and quasi-harmonic analysis for entropic calculations. Finally, a few of the fundamental findings that have resulted from these methods are discussed, such as the role of configurational entropy and solvent in intermolecular interactions, along with selected results of the model system T4 lysozyme to illustrate potential and current limitations of these methods.
Molecular recognition plays a central role in biochemical processes. Although well studied, understanding the mechanisms of recognition is inherently difficult due to the range of potential interactions, the molecular rearrangement associated with binding, and the time and length scales involved. Computational methods have the potential for not only complementing experiments that have been performed, but also in guiding future ones through their predictive abilities. In this review, we discuss how molecular dynamics (MD) simulations may be used in advancing our understanding of the thermodynamics that drive biomolecular recognition. We begin with a brief review of the statistical mechanics that form a basis for these methods. This is followed by a description of some of the most commonly used methods: thermodynamic pathways employing alchemical transformations and potential of mean force calculations, along with end-point calculations for free energy differences, and harmonic and quasi-harmonic analysis for entropic calculations. Finally, a few of the fundamental findings that have resulted from these methods are discussed, such as the role of configurational entropy and solvent in intermolecular interactions, along with selected results of the model system T4 lysozyme to illustrate potential and current limitations of these methods.
Adaptive Finite Element Modeling Techniques for the Poisson-Boltzmann EquationCommunications in Computational Physics, Vol. 11, pp. 179-214 (2012) [PubMed 21949541]
We consider the design of an effective and reliable adaptive finite element method (AFEM) for the nonlinear Poisson-Boltzmann equation (PBE). We first examine the two-term regularization technique for the continuous problem recently proposed by Chen, Holst, and Xu based on the removal of the singular electrostatic potential inside biomolecules; this technique made possible the development of the first complete solution and approximation theory for the Poisson-Boltzmann equation, the first provably convergent discretization, and also allowed for the development of a provably convergent AFEM. However, in practical implementation, this two-term regularization exhibits numerical instability. Therefore, we examine a variation of this regularization technique which can be shown to be less susceptible to such instability. We establish a priori estimates and other basic results for the continuous regularized problem, as well as for Galerkin finite element approximations. We show that the new approach produces regularized continuous and discrete problems with the same mathematical advantages of the original regularization. We then design an AFEM scheme for the new regularized problem, and show that the resulting AFEM scheme is accurate and reliable, by proving a contraction result for the error. This result, which is one of the first results of this type for nonlinear elliptic problems, is based on using continuous and discrete a priori L1 estimates to establish quasi-orthogonality. To provide a high-quality geometric model as input to the AFEM algorithm, we also describe a class of feature-preserving adaptive mesh generation algorithms designed specifically for constructing meshes of biomolecular structures, based on the intrinsic local structure tensor of the molecular surface. All of the algorithms described in the article are implemented in the Finite Element Toolkit (FETK), developed and maintained at UCSD. The stability advantages of the new regularization scheme are demonstrated with FETK through comparisons with the original regularization approach for a model problem. The convergence and accuracy of the overall AFEM algorithm is also illustrated by numerical approximation of electrostatic solvation energy for an insulin protein.
We consider the design of an effective and reliable adaptive finite element method (AFEM) for the nonlinear Poisson-Boltzmann equation (PBE). We first examine the two-term regularization technique for the continuous problem recently proposed by Chen, Holst, and Xu based on the removal of the singular electrostatic potential inside biomolecules; this technique made possible the development of the first complete solution and approximation theory for the Poisson-Boltzmann equation, the first provably convergent discretization, and also allowed for the development of a provably convergent AFEM. However, in practical implementation, this two-term regularization exhibits numerical instability. Therefore, we examine a variation of this regularization technique which can be shown to be less susceptible to such instability. We establish a priori estimates and other basic results for the continuous regularized problem, as well as for Galerkin finite element approximations. We show that the new approach produces regularized continuous and discrete problems with the same mathematical advantages of the original regularization. We then design an AFEM scheme for the new regularized problem, and show that the resulting AFEM scheme is accurate and reliable, by proving a contraction result for the error. This result, which is one of the first results of this type for nonlinear elliptic problems, is based on using continuous and discrete a priori L1 estimates to establish quasi-orthogonality. To provide a high-quality geometric model as input to the AFEM algorithm, we also describe a class of feature-preserving adaptive mesh generation algorithms designed specifically for constructing meshes of biomolecular structures, based on the intrinsic local structure tensor of the molecular surface. All of the algorithms described in the article are implemented in the Finite Element Toolkit (FETK), developed and maintained at UCSD. The stability advantages of the new regularization scheme are demonstrated with FETK through comparisons with the original regularization approach for a model problem. The convergence and accuracy of the overall AFEM algorithm is also illustrated by numerical approximation of electrostatic solvation energy for an insulin protein.
Introduction to Molecular Dynamics: Theory and Applications in Biomolecular ModelingIn "Computational Modeling of Biological Systems: From Molecules to Pathways", N.V. Dokholyan (Ed.), Ch. 1, Springer-Verlag (2012)
Independent-Trajectory Thermodynamic Integration: A Practical Guide to Protein-Drug Binding Free Energy Calculations Using Distributed ComputingIn "Computational Drug Discovery and Design", R. Baron (Ed.), Springer Series in Methods in Molecular Biology. Humana Press, USA, pp. 469-486 (2012) [PubMed 22183552]
The Independent-Trajectory Thermodynamic Integration (IT-TI) approach for free energy calculation with distributed computing is described. IT-TI utilizes diverse conformational sampling obtained from multiple, independent simulations to obtain more reliable free energy estimates compared to single TI predictions. The latter may significantly under- or over-estimate the binding free energy due to finite sampling. We exemplify the advantages of the IT-TI approach using two distinct cases of protein-ligand binding. In both cases, IT-TI yields distributions of absolute binding free energy estimates that are remarkably centered on the target experimental values. Alternative protocols for the practical and general application of IT-TI calculations are investigated. We highlight a protocol that maximizes predictive power and computational efficiency.
The Independent-Trajectory Thermodynamic Integration (IT-TI) approach for free energy calculation with distributed computing is described. IT-TI utilizes diverse conformational sampling obtained from multiple, independent simulations to obtain more reliable free energy estimates compared to single TI predictions. The latter may significantly under- or over-estimate the binding free energy due to finite sampling. We exemplify the advantages of the IT-TI approach using two distinct cases of protein-ligand binding. In both cases, IT-TI yields distributions of absolute binding free energy estimates that are remarkably centered on the target experimental values. Alternative protocols for the practical and general application of IT-TI calculations are investigated. We highlight a protocol that maximizes predictive power and computational efficiency.
Guide to Virtual Screening: Application to the Akt Phosphatase PHLPPIn "Computational Drug Discovery and Design", R. Baron (Ed.), Springer Series in Methods in Molecular Biology. Humana Press, USA, pp. 561-573 (2012) [PubMed 22183558]
We present an example-based description of virtual screening (VS) techniques used to identify new regulators of the Akt phosphatase PHLPP (PH domain Leucine repeat Protein Phosphatase). This enzyme opposes the effects of two kinases, Akt and PKC, which play a major role in cell growth and survival. Therefore, PHLPP is a potential therapeutic target in pathophysiologies where these pathways are either repressed, such as in diabetes and cardiovascular diseases, or over-activated as in cancer. To the best of our knowledge, no PHLPP inhibitors have been reported so far in the literature. In this study, we used a combination of chemical and virtual screening techniques that led to the identification of a number of inhibiting compounds with diverse scaffolds. These compounds bind PHLPP and inhibit cell death when tested in cellular assays. We employed GLIDE docking software to screen a library of more than 40,000 compounds selected from the NCI open depository (250,000 compounds) by similarity searches. We compare the efficiency at which we determined binding compounds from the chemical screen, and compare enrichment factors of the virtually discovered compounds over chemical screening.
We present an example-based description of virtual screening (VS) techniques used to identify new regulators of the Akt phosphatase PHLPP (PH domain Leucine repeat Protein Phosphatase). This enzyme opposes the effects of two kinases, Akt and PKC, which play a major role in cell growth and survival. Therefore, PHLPP is a potential therapeutic target in pathophysiologies where these pathways are either repressed, such as in diabetes and cardiovascular diseases, or over-activated as in cancer. To the best of our knowledge, no PHLPP inhibitors have been reported so far in the literature. In this study, we used a combination of chemical and virtual screening techniques that led to the identification of a number of inhibiting compounds with diverse scaffolds. These compounds bind PHLPP and inhibit cell death when tested in cellular assays. We employed GLIDE docking software to screen a library of more than 40,000 compounds selected from the NCI open depository (250,000 compounds) by similarity searches. We compare the efficiency at which we determined binding compounds from the chemical screen, and compare enrichment factors of the virtually discovered compounds over chemical screening.
A Molecular Dynamics Ensemble-Based Approach for the Mapping of Druggable Binding SitesIn "Computational Drug Discovery and Design", R. Baron (Ed.), Springer Series in Methods in Molecular Biology, Humana Press, USA, pp. 3-12 (2012) [PubMed 22183526]
An expanding repertoire of "allosteric" drugs is revealing that structure-based drug design (SBDD) is not restricted to the "active site" of the target protein. Such compounds have been shown to bind distant regions of the protein topography, potentially providing higher levels of target specificity, reduced toxicity and access to new regions of chemical space. Unfortunately, the location of such allosteric pockets is not obvious in the absence of a bound crystal structure and the ability to predict their presence would be useful in the discovery of novel therapies. Here, we describe a method for the prediction of "druggable" binding sites that takes protein flexibility into account through the use of molecular dynamics (MD) simulation. By using a dynamic representation of the target, we are able to sample multiple protein conformations that may expose new drug-binding surfaces. We perform a fragment-based mapping analysis of individual structures in the MD ensemble using the FTMAP algorithm and then rank the most prolific probe-binding protein residues to determine potential "hot-spots" for further examination. This approach has recently been applied to a pair of human G-protein-coupled receptors (GPCRs), resulting in the detection of five potential allosteric sites.
An expanding repertoire of "allosteric" drugs is revealing that structure-based drug design (SBDD) is not restricted to the "active site" of the target protein. Such compounds have been shown to bind distant regions of the protein topography, potentially providing higher levels of target specificity, reduced toxicity and access to new regions of chemical space. Unfortunately, the location of such allosteric pockets is not obvious in the absence of a bound crystal structure and the ability to predict their presence would be useful in the discovery of novel therapies. Here, we describe a method for the prediction of "druggable" binding sites that takes protein flexibility into account through the use of molecular dynamics (MD) simulation. By using a dynamic representation of the target, we are able to sample multiple protein conformations that may expose new drug-binding surfaces. We perform a fragment-based mapping analysis of individual structures in the MD ensemble using the FTMAP algorithm and then rank the most prolific probe-binding protein residues to determine potential "hot-spots" for further examination. This approach has recently been applied to a pair of human G-protein-coupled receptors (GPCRs), resulting in the detection of five potential allosteric sites.
Accelerated Molecular Dynamics in Computational Drug DesignIn "Computational Drug Discovery and Design", R. Baron (Ed.), Springer Series in Methods in Molecular Biology, Humana Press, USA, pp. 515-524 (2012) [PubMed 22183555]
The method of accelerated molecular dynamics (aMD) has been shown to increase the rate of phase-space sampling in biomolecular simulations. In this chapter, we discuss the theory behind aMD and describe the implementation of two versions: dual-boost and selective aMD. Each method has its practical advantages: dual-boost aMD is useful for increasing sampling of global conformational motions while selective aMD can improve the rate of convergence of free energy calculations. Special emphasis is placed on the use of these methods in computer-aided drug design, and the example of oseltamivir binding to neuraminidase is highlighted for both cases.
The method of accelerated molecular dynamics (aMD) has been shown to increase the rate of phase-space sampling in biomolecular simulations. In this chapter, we discuss the theory behind aMD and describe the implementation of two versions: dual-boost and selective aMD. Each method has its practical advantages: dual-boost aMD is useful for increasing sampling of global conformational motions while selective aMD can improve the rate of convergence of free energy calculations. Special emphasis is placed on the use of these methods in computer-aided drug design, and the example of oseltamivir binding to neuraminidase is highlighted for both cases.
On the Use of Molecular Dynamics Receptor Conformations for Virtual ScreeningIn "Computational Drug Discovery and Design", R. Baron (Ed.), Springer Series in Methods in Molecular Biology, Humana Press, USA, pp. 93-103 (2012) [PubMed 22183532]
Receptors are inherently dynamic and this flexibility is important to consider when constructing a model of molecular association. Conformations from molecular dynamics simulations, a well-established method for examining protein dynamics, can be used in virtual screening to account for flexibility in structure-based drug discovery. Different receptor configurations influence docking results. Molecular dynamics simulations can provide snapshots that improve virtual screening predictive power over known crystal structures, most likely as a result of sampling more relevant receptor conformations. Here we highlight some details and nuances of using such snapshots and evaluating them for predictive performance.
Receptors are inherently dynamic and this flexibility is important to consider when constructing a model of molecular association. Conformations from molecular dynamics simulations, a well-established method for examining protein dynamics, can be used in virtual screening to account for flexibility in structure-based drug discovery. Different receptor configurations influence docking results. Molecular dynamics simulations can provide snapshots that improve virtual screening predictive power over known crystal structures, most likely as a result of sampling more relevant receptor conformations. Here we highlight some details and nuances of using such snapshots and evaluating them for predictive performance.
Hydrophobic Association and Volume-confined Water MoleculesIn "Protein-ligand Interactions", H. Gohlke (Ed.), Methods and Principles in Medicinal Chemistry Series, Wiley-VCH Publishers (2012)
Mathematical and Numerical Aspects of the Adaptive Fast Multipole Poisson-Boltzmann SolverCommunications in Computational Physics, Vol. 13, Issue 1, pp. 107-128 (2012)
This paper summarizes the mathematical and numerical theories and computational elements of the adaptive fast multipole Poisson-Boltzmann (AFMPB) solver. We introduce and discuss the following components in order: the Poisson-Boltzmann model, boundary integral equation reformulation, surface mesh generation, the node-patch discretization approach, Krylov iterative methods, the new version of fast multipole methods (FMMs), and a dynamic prioritization technique for scheduling parallel operations. For each component, we also remark on feasible approaches for further improvements in efficiency, accuracy and applicability of the AFMPB solver to large-scale long-time molecular dynamics simulations. The potential of the solver is demonstrated with preliminary numerical results.
This paper summarizes the mathematical and numerical theories and computational elements of the adaptive fast multipole Poisson-Boltzmann (AFMPB) solver. We introduce and discuss the following components in order: the Poisson-Boltzmann model, boundary integral equation reformulation, surface mesh generation, the node-patch discretization approach, Krylov iterative methods, the new version of fast multipole methods (FMMs), and a dynamic prioritization technique for scheduling parallel operations. For each component, we also remark on feasible approaches for further improvements in efficiency, accuracy and applicability of the AFMPB solver to large-scale long-time molecular dynamics simulations. The potential of the solver is demonstrated with preliminary numerical results.
Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics SimulationsJournal of Chemical Theory and Computation, 8 (1), pp 17–23 (2012) [PubMed 22241967]
Molecular dynamics (MD) is one of the most common tools in computational chemistry. Recently, our group has employed accelerated molecular dynamics (aMD) to improve the conformational sampling over conventional molecular dynamics techniques. In the original aMD implementation, sampling is greatly improved by raising energy wells below a predefined energy level. Recently, our group presented an alternative aMD implementation where simulations are accelerated by lowering energy barriers of the potential energy surface. When coupled with thermodynamic integration simulations, this implementation showed very promising results. However, when applied to large systems, such as proteins, the simulation tends to be biased to high energy regions of the potential landscape. The reason for this behavior lies in the boost equation used since the highest energy barriers are dramatically more affected than the lower ones. To address this issue, in this work, we present a new boost equation that prevents oversampling of unfavorable high energy conformational states. The new boost potential provides not only better recovery of statistics throughout the simulation but also enhanced sampling of statistically relevant regions in explicit solvent MD simulations.
Molecular dynamics (MD) is one of the most common tools in computational chemistry. Recently, our group has employed accelerated molecular dynamics (aMD) to improve the conformational sampling over conventional molecular dynamics techniques. In the original aMD implementation, sampling is greatly improved by raising energy wells below a predefined energy level. Recently, our group presented an alternative aMD implementation where simulations are accelerated by lowering energy barriers of the potential energy surface. When coupled with thermodynamic integration simulations, this implementation showed very promising results. However, when applied to large systems, such as proteins, the simulation tends to be biased to high energy regions of the potential landscape. The reason for this behavior lies in the boost equation used since the highest energy barriers are dramatically more affected than the lower ones. To address this issue, in this work, we present a new boost equation that prevents oversampling of unfavorable high energy conformational states. The new boost potential provides not only better recovery of statistics throughout the simulation but also enhanced sampling of statistically relevant regions in explicit solvent MD simulations.
Computer-aided Drug Discovery: Two Antiviral Drugs for HIV/AIDSIn "Innovations in Biomolecular Modelling and Simulations", Vol. 2, Tamar Schlick (Ed.), Royal Society of Chemistry (2012)
Level-Set Variational Implicit-Solvent Modeling of Biomolecules with the Coulomb-Field ApproximationJournal of Chemical Theory and Computation, 8 (2), pp 386–397 (2012) [PubMed 22346739]
Central in the variational implicit-solvent model (VISM) Dzubiella, Swanson, and McCammon Phys. Rev. Lett.2006, 96, 087802 and J. Chem. Phys.2006, 124, 084905 of molecular solvation is a mean-field free-energy functional of all possible solute–solvent interfaces or dielectric boundaries. Such a functional can be minimized numerically by a level-set method to determine stable equilibrium conformations and solvation free energies. Applications to nonpolar systems have shown that the level-set VISM is efficient and leads to qualitatively and often quantitatively correct results. In particular, it is capable of capturing capillary evaporation in hydrophobic confinement and corresponding multiple equilibrium states as found in molecular dynamics (MD) simulations. In this work, we introduce into the VISM the Coulomb-field approximation of the electrostatic free energy. Such an approximation is a volume integral over an arbitrary shaped solvent region, requiring no solutions to any partial differential equations. With this approximation, we obtain the effective boundary force and use it as the “normal velocity” in the level-set relaxation. We test the new approach by calculating solvation free energies and potentials of mean force for small and large molecules, including the two-domain protein BphC. Our results reveal the importance of coupling polar and nonpolar interactions in the underlying molecular systems. In particular, dehydration near the domain interface of BphC subunits is found to be highly sensitive to local electrostatic potentials as seen in previous MD simulations. This is a first step toward capturing the complex protein dehydration process by an implicit-solvent approach.
Central in the variational implicit-solvent model (VISM) Dzubiella, Swanson, and McCammon Phys. Rev. Lett.2006, 96, 087802 and J. Chem. Phys.2006, 124, 084905 of molecular solvation is a mean-field free-energy functional of all possible solute–solvent interfaces or dielectric boundaries. Such a functional can be minimized numerically by a level-set method to determine stable equilibrium conformations and solvation free energies. Applications to nonpolar systems have shown that the level-set VISM is efficient and leads to qualitatively and often quantitatively correct results. In particular, it is capable of capturing capillary evaporation in hydrophobic confinement and corresponding multiple equilibrium states as found in molecular dynamics (MD) simulations. In this work, we introduce into the VISM the Coulomb-field approximation of the electrostatic free energy. Such an approximation is a volume integral over an arbitrary shaped solvent region, requiring no solutions to any partial differential equations. With this approximation, we obtain the effective boundary force and use it as the “normal velocity” in the level-set relaxation. We test the new approach by calculating solvation free energies and potentials of mean force for small and large molecules, including the two-domain protein BphC. Our results reveal the importance of coupling polar and nonpolar interactions in the underlying molecular systems. In particular, dehydration near the domain interface of BphC subunits is found to be highly sensitive to local electrostatic potentials as seen in previous MD simulations. This is a first step toward capturing the complex protein dehydration process by an implicit-solvent approach.
AutoClickChem: Click Chemistry in SilicoPLoS Computational Biology, Vol. 8, Issue 3, e1002397 (2012) [PubMed 22438795]
Academic researchers and many in industry often lack the financial resources available to scientists working in “big pharma.” High costs include those associated with high-throughput screening and chemical synthesis. In order to address these challenges, many researchers have in part turned to alternate methodologies. Virtual screening, for example, often substitutes for high-throughput screening, and click chemistry ensures that chemical synthesis is fast, cheap, and comparatively easy. Though both in silico screening and click chemistry seek to make drug discovery more feasible, it is not yet routine to couple these two methodologies. We here present a novel computer algorithm, called AutoClickChem, capable of performing many click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compound models for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Additionally, in silico modeling of click-chemistry products may prove useful in rational drug design and drug optimization. AutoClickChem is based on the pymolecule toolbox, a framework that may facilitate the development of future python-based programs that require the manipulation of molecular models. Both the pymolecule toolbox and AutoClickChem are released under the GNU General Public License version 3 and are available for download from http://autoclickchem.ucsd.edu">http://autoclickchem.ucsd.edu.
Academic researchers and many in industry often lack the financial resources available to scientists working in “big pharma.” High costs include those associated with high-throughput screening and chemical synthesis. In order to address these challenges, many researchers have in part turned to alternate methodologies. Virtual screening, for example, often substitutes for high-throughput screening, and click chemistry ensures that chemical synthesis is fast, cheap, and comparatively easy. Though both in silico screening and click chemistry seek to make drug discovery more feasible, it is not yet routine to couple these two methodologies. We here present a novel computer algorithm, called AutoClickChem, capable of performing many click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compound models for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Additionally, in silico modeling of click-chemistry products may prove useful in rational drug design and drug optimization. AutoClickChem is based on the pymolecule toolbox, a framework that may facilitate the development of future python-based programs that require the manipulation of molecular models. Both the pymolecule toolbox and AutoClickChem are released under the GNU General Public License version 3 and are available for download from http://autoclickchem.ucsd.edu">http://autoclickchem.ucsd.edu.
Accelerated Molecular Dynamics: Theory, Implementation and ApplicationIn "Proceedings of the 2012 Conference on Theory and Applications of Computational Chemistry", E. Clementi, Ed., American Institute of Physics (2012)
Simulations of the p97 complex suggest novel conformational states of hydrolysis intermediatesProtein Science, Vol. 21, Issue 4, pp. 475-486 (2012) [PubMed 22238181]
The vitally important AAA protein p97 is involved in cellular functions ranging from replication to degradation of misfolded proteins and has recently been proposed as a novel chemotherapeutic target. p97 is a large molecular machine that has been shown to hexamerize in vitro, with each monomer consisting of an N-domain responsible for binding to effector proteins and two AAA repeats (D1 and D2). However, structural studies are inconclusive or in disagreement with one another on several important features such as the locations of the N domains, the relative orientations of the D1 and D2 rings, and the dimensions of the central pore. Here, we present atomic-scale simulations of the p97 hexamer in the pre-hydrolysis, transition, and post-hydrolysis states. To improve the agreement between low- and high-resolution experimental studies, we first use a biased simulation technique, molecular dynamics flexible fitting (MDFF), to improve the correlation between the structures described in these experiments. We follow this with extended, classical molecular dynamics simulations, which not only show that structures generated in the MDFF phase are stable, but reveal insights into the dynamics important to each state. Simulation results suggest a hybrid model for hydrolysis, in which the N and D2 domains are dynamic while the D1 domains are relatively static, salt bridges stabilize the position of the N-domains in the pre-hydrolysis state, and the rings formed by D1 and D2 rotate relative to one another.
The vitally important AAA protein p97 is involved in cellular functions ranging from replication to degradation of misfolded proteins and has recently been proposed as a novel chemotherapeutic target. p97 is a large molecular machine that has been shown to hexamerize in vitro, with each monomer consisting of an N-domain responsible for binding to effector proteins and two AAA repeats (D1 and D2). However, structural studies are inconclusive or in disagreement with one another on several important features such as the locations of the N domains, the relative orientations of the D1 and D2 rings, and the dimensions of the central pore. Here, we present atomic-scale simulations of the p97 hexamer in the pre-hydrolysis, transition, and post-hydrolysis states. To improve the agreement between low- and high-resolution experimental studies, we first use a biased simulation technique, molecular dynamics flexible fitting (MDFF), to improve the correlation between the structures described in these experiments. We follow this with extended, classical molecular dynamics simulations, which not only show that structures generated in the MDFF phase are stable, but reveal insights into the dynamics important to each state. Simulation results suggest a hybrid model for hydrolysis, in which the N and D2 domains are dynamic while the D1 domains are relatively static, salt bridges stabilize the position of the N-domains in the pre-hydrolysis state, and the rings formed by D1 and D2 rotate relative to one another.
Multiscale Continuum Modeling and Simulation of Biological Processes: From Molecular Electro-diffusion to Sub-Cellular Signaling TransductionComputational Science & Discovery, Vol. 5, Issue 1, pp. 475-486 (2012) [PubMed 23505398]
This paper presents a brief review of multi-scale modeling at the molecular to cellular scale, with new results for heart muscle cells. A finite element-based simulation package (SMOL) was used to investigate the signaling transduction at molecular and sub-cellular scales ( http://mccammon.ucsd.edu/smol/ , http://FETK.org ) by numerical solution of the time-dependent Smoluchowski equations and a reaction-diffusion system. At the molecular scale, SMOL has yielded experimentally validated estimates of the diffusion-limited association rates for the binding of acetylcholine to mouse acetylcholinesterase using crystallographic structural data. The predicted rate constants exhibit increasingly delayed steady-state times, with increasing ionic strength, and demonstrate the role of an enzyme's electrostatic potential in influencing ligand binding. At the sub-cellular scale, an extension of SMOL solves a nonlinear, reaction-diffusion system describing Ca 2+ ligand buffering and diffusion in experimentally derived rodent ventricular myocyte geometries. Results reveal the important role of mobile and stationary Ca 2+ buffers, including Ca 2+ indicator dye. We found that alterations in Ca 2+ -binding and dissociation rates of troponin C (TnC) and total TnC concentration modulate sub-cellular Ca 2+ signals. The model predicts that reduced off-rate in the whole troponin complex (TnC, TnI, TnT) versus reconstructed thin filaments (Tn, Tm, actin) alters cytosolic Ca 2+ dynamics under control conditions or in disease-linked TnC mutations. The ultimate goal of these studies is to develop scalable methods and theories for the integration of molecular-scale information into simulations of cellular-scale systems.
This paper presents a brief review of multi-scale modeling at the molecular to cellular scale, with new results for heart muscle cells. A finite element-based simulation package (SMOL) was used to investigate the signaling transduction at molecular and sub-cellular scales ( http://mccammon.ucsd.edu/smol/ , http://FETK.org ) by numerical solution of the time-dependent Smoluchowski equations and a reaction-diffusion system. At the molecular scale, SMOL has yielded experimentally validated estimates of the diffusion-limited association rates for the binding of acetylcholine to mouse acetylcholinesterase using crystallographic structural data. The predicted rate constants exhibit increasingly delayed steady-state times, with increasing ionic strength, and demonstrate the role of an enzyme's electrostatic potential in influencing ligand binding. At the sub-cellular scale, an extension of SMOL solves a nonlinear, reaction-diffusion system describing Ca 2+ ligand buffering and diffusion in experimentally derived rodent ventricular myocyte geometries. Results reveal the important role of mobile and stationary Ca 2+ buffers, including Ca 2+ indicator dye. We found that alterations in Ca 2+ -binding and dissociation rates of troponin C (TnC) and total TnC concentration modulate sub-cellular Ca 2+ signals. The model predicts that reduced off-rate in the whole troponin complex (TnC, TnI, TnT) versus reconstructed thin filaments (Tn, Tm, actin) alters cytosolic Ca 2+ dynamics under control conditions or in disease-linked TnC mutations. The ultimate goal of these studies is to develop scalable methods and theories for the integration of molecular-scale information into simulations of cellular-scale systems.
Thermodynamic integration to predict host-guest binding affinitiesJournal of Computer-Aided Molecular Design, Vol. 26, Issue 5, pp. 569-576 (2012) [PubMed 22350568]
An alchemical free energy method with explicit solvent molecular dynamics simulations was applied as part of the blind prediction contest SAMPL3 to calculate binding free energies for seven guests to an acyclic cucurbit-nuril host. The predictions included determination of protonation states for both host and guests, docking pose generation, and binding free energy calculations using thermodynamic integration. We found a root mean square error (RMSE) of Formula: see text from the reference experimental results, with an R (2) correlation of 0.51. The agreement with experiment for the largest contributor to this error, guest 6, is improved by Formula: see text when a periodicity-induced free energy correction is applied. The corrections for the other ligands were significantly smaller, and altogether the RMSE was reduced by Formula: see text. We link properties of the host-guest systems during simulation to errors in the computed free energies. Overall, we show that charged host-guest systems studied here, initialized in unconfirmed docking poses, present a challenge to accurate alchemical simulation methods.
An alchemical free energy method with explicit solvent molecular dynamics simulations was applied as part of the blind prediction contest SAMPL3 to calculate binding free energies for seven guests to an acyclic cucurbit-nuril host. The predictions included determination of protonation states for both host and guests, docking pose generation, and binding free energy calculations using thermodynamic integration. We found a root mean square error (RMSE) of Formula: see text from the reference experimental results, with an R (2) correlation of 0.51. The agreement with experiment for the largest contributor to this error, guest 6, is improved by Formula: see text when a periodicity-induced free energy correction is applied. The corrections for the other ligands were significantly smaller, and altogether the RMSE was reduced by Formula: see text. We link properties of the host-guest systems during simulation to errors in the computed free energies. Overall, we show that charged host-guest systems studied here, initialized in unconfirmed docking poses, present a challenge to accurate alchemical simulation methods.
Dynamics and calcium association to the N-terminal regulatory domain of human cardiac Troponin C: A multi-scale computational studyJ. Phys. Chem. B, Vol. 116, Issue 29, pp. 8449-8459 (2012) [PubMed 22329450]
Troponin C (TnC) is an important regulatory molecule in cardiomyocytes. Calcium binding to site II in TnC initiates a series of molecular events that result in muscle contraction. The most direct change upon Ca(2+) binding is an opening motion of the molecule that exposes a hydrophobic patch on the surface allowing for Troponin I to bind. Molecular dynamics simulations were used to elucidate the dynamics of this crucial protein in three different states: apo, Ca(2+)-bound, and Ca(2+)-TnI-bound. Dynamics between the states are compared, and the Ca(2+)-bound system is investigated for opening motions. On the basis of the simulations, NMR chemical shifts and order parameters are calculated and compared with experimental observables. Agreement indicates that the simulations sample the relevant dynamics of the system. Brownian dynamics simulations are used to investigate the calcium association of TnC. We find that calcium binding gives rise to correlative motions involving the EF hand and collective motions conducive of formation of the TnI-binding interface. We furthermore indicate the essential role of electrostatic steering in facilitating diffusion-limited binding of Ca(2+).
Troponin C (TnC) is an important regulatory molecule in cardiomyocytes. Calcium binding to site II in TnC initiates a series of molecular events that result in muscle contraction. The most direct change upon Ca(2+) binding is an opening motion of the molecule that exposes a hydrophobic patch on the surface allowing for Troponin I to bind. Molecular dynamics simulations were used to elucidate the dynamics of this crucial protein in three different states: apo, Ca(2+)-bound, and Ca(2+)-TnI-bound. Dynamics between the states are compared, and the Ca(2+)-bound system is investigated for opening motions. On the basis of the simulations, NMR chemical shifts and order parameters are calculated and compared with experimental observables. Agreement indicates that the simulations sample the relevant dynamics of the system. Brownian dynamics simulations are used to investigate the calcium association of TnC. We find that calcium binding gives rise to correlative motions involving the EF hand and collective motions conducive of formation of the TnI-binding interface. We furthermore indicate the essential role of electrostatic steering in facilitating diffusion-limited binding of Ca(2+).
Comparative Molecular Dynamics Simulations of the Antimicrobial Peptide CM15 in Model Lipid BilayersBiochim Biophys Acta, Vol. 1818, Issue 5, pp. 1402-1409 (2012) [PubMed 22387432]
We report altogether 3-μs molecular dynamics (MD) simulations of the antimicrobial peptide CM15 to systematically investigate its interaction with two model lipid bilayers, pure POPC and mixed POPG:POPC (1:2). Starting with either an α-helical or a random-coil conformation, CM15 is found to insert into both bilayers. Peptide-lipid interaction is stronger with the anionic POPG:POPC than the zwitterionic POPC, which is largely attributed to the electrostatic attraction between CM15 and the negatively charged POPG. Simulations initiated with CM15 as a random coil allowed us to study peptide folding at the lipid-water interface. Interestingly, CM15 folding appears to be faster in POPC than POPG:POPC, which may be explained by a lower activation energy barrier of structural rearrangement in the former system. Our data also suggest that compared with the random-coil conformation, CM15 in a pre-folded α-helix has significantly reduced interactions with the lipids, indicating that peptide initial structures may bias the simulation results considerably on the 100-ns timescale. The implications of this result should be considered when preparing and interpreting future AMP simulations
We report altogether 3-μs molecular dynamics (MD) simulations of the antimicrobial peptide CM15 to systematically investigate its interaction with two model lipid bilayers, pure POPC and mixed POPG:POPC (1:2). Starting with either an α-helical or a random-coil conformation, CM15 is found to insert into both bilayers. Peptide-lipid interaction is stronger with the anionic POPG:POPC than the zwitterionic POPC, which is largely attributed to the electrostatic attraction between CM15 and the negatively charged POPG. Simulations initiated with CM15 as a random coil allowed us to study peptide folding at the lipid-water interface. Interestingly, CM15 folding appears to be faster in POPC than POPG:POPC, which may be explained by a lower activation energy barrier of structural rearrangement in the former system. Our data also suggest that compared with the random-coil conformation, CM15 in a pre-folded α-helix has significantly reduced interactions with the lipids, indicating that peptide initial structures may bias the simulation results considerably on the 100-ns timescale. The implications of this result should be considered when preparing and interpreting future AMP simulations
Modeling Cardiac Calcium Sparks in a Three-Dimensional Reconstruction of a Calcium Release UnitJ. Physiol, Vol. 590, Issue 18, pp. 4403-4422 (2012) [PubMed 22495592]
Triggered release of Ca2+ from an individual sarcoplasmic reticulum (SR) Ca2+ release unit (CRU) is the fundamental event of cardiac excitation-contraction coupling, and spontaneous release events (sparks) are the major contributor to diastolic Ca2+ leak in cardiomyocytes. Previous model studies have predicted that the duration and magnitude of the spark is determined by the local CRU geometry, as well as the localization and density of Ca2+ handling proteins. We have created a detailed computational model of a CRU, and developed novel tools to generate the computational geometry from electron tomographic images. Ca2+ diffusion was modeled within the SR and the cytosol to examine the effects of localization and density of the Na+ /Ca2+ exchanger, Sarco/endoplasmic reticulum Ca2+ ATPase 2 (SERCA), and calsequestrin on spark dynamics. We reconcile previous model predictions of approximately 90 % local Ca2+ depletion in junctional SR, with experimental reports of about 40 %. This analysis supports the hypothesis that dye kinetics and optical averaging effects can have a significant impact on measures of spark dynamics. Our model also predicts that distributing calsequestrin within non-junctional Z-disc SR compartments, in addition to the junctional compartment, prolongs spark release time as reported by Fluo5. By pumping Ca2+ back into the SR during a release, SERCA is able to prolong a Ca2+ spark, and this may contribute to SERCA-dependent changes in Ca2+ -wave speed. Finally, we show that including the Na+ /Ca2+ exchanger inside the dyadic cleft does not alter local Ca2+ during a spark.
Triggered release of Ca2+ from an individual sarcoplasmic reticulum (SR) Ca2+ release unit (CRU) is the fundamental event of cardiac excitation-contraction coupling, and spontaneous release events (sparks) are the major contributor to diastolic Ca2+ leak in cardiomyocytes. Previous model studies have predicted that the duration and magnitude of the spark is determined by the local CRU geometry, as well as the localization and density of Ca2+ handling proteins. We have created a detailed computational model of a CRU, and developed novel tools to generate the computational geometry from electron tomographic images. Ca2+ diffusion was modeled within the SR and the cytosol to examine the effects of localization and density of the Na+ /Ca2+ exchanger, Sarco/endoplasmic reticulum Ca2+ ATPase 2 (SERCA), and calsequestrin on spark dynamics. We reconcile previous model predictions of approximately 90 % local Ca2+ depletion in junctional SR, with experimental reports of about 40 %. This analysis supports the hypothesis that dye kinetics and optical averaging effects can have a significant impact on measures of spark dynamics. Our model also predicts that distributing calsequestrin within non-junctional Z-disc SR compartments, in addition to the junctional compartment, prolongs spark release time as reported by Fluo5. By pumping Ca2+ back into the SR during a release, SERCA is able to prolong a Ca2+ spark, and this may contribute to SERCA-dependent changes in Ca2+ -wave speed. Finally, we show that including the Na+ /Ca2+ exchanger inside the dyadic cleft does not alter local Ca2+ during a spark.
Nucleotide dependent mechanism of Get3 as elucidated from free energy calculationsProc. Natl. Acad. Sci. USA Vol. 109. No. 20 7759-7764 (2012) [PubMed 22547793]
The unique topology of tail-anchored (TA) proteins precludes them from utilizing the well-studied cotranslational translocation mechanism of most transmembrane proteins, forcing them into a distinct, posttranslational pathway. In yeast, this process is the guided entry of TA-proteins (GET) pathway, which utilizes a combination of cytosolic and transmembrane proteins to identify a TA protein, transfer it, and insert it into the endoplasmic reticulum membrane. At the center of this mechanism is the Get3 homodimer, which transfers a TA protein between the two GET phases by leveraging energy gained in ATP binding and hydrolysis to undergo significant structural changes from “open” to “closed” conformations. We present all-atom molecular dynamics simulations of Get3 in multiple nucleotide states, and through rigorous potential of mean force calculations, compute the free energy landscape of the Get3 opening/closing pathway. Results agree well with experiments on the nucleotide bias of Get3 open and closed structures in the crystallographically observed no-nucleotide, two ATP, and two ADP states, and also reveal their populations in the asymmetric one
ATP and one ADP cases. Structures also compare well with the recently observed “semiopen” conformation and suggest that Get3 may sample this state free in solution and not just when bound to Get1, as observed in experiments. Finally, we present evidence for a unique, “wide-open” conformation of Get3. These calculations describe the nucleotide-dependent thermodynamics of Get3 in solution, and improve our understanding of its mechanism in each phase of the GET cycle.
The unique topology of tail-anchored (TA) proteins precludes them from utilizing the well-studied cotranslational translocation mechanism of most transmembrane proteins, forcing them into a distinct, posttranslational pathway. In yeast, this process is the guided entry of TA-proteins (GET) pathway, which utilizes a combination of cytosolic and transmembrane proteins to identify a TA protein, transfer it, and insert it into the endoplasmic reticulum membrane. At the center of this mechanism is the Get3 homodimer, which transfers a TA protein between the two GET phases by leveraging energy gained in ATP binding and hydrolysis to undergo significant structural changes from “open” to “closed” conformations. We present all-atom molecular dynamics simulations of Get3 in multiple nucleotide states, and through rigorous potential of mean force calculations, compute the free energy landscape of the Get3 opening/closing pathway. Results agree well with experiments on the nucleotide bias of Get3 open and closed structures in the crystallographically observed no-nucleotide, two ATP, and two ADP states, and also reveal their populations in the asymmetric one
ATP and one ADP cases. Structures also compare well with the recently observed “semiopen” conformation and suggest that Get3 may sample this state free in solution and not just when bound to Get1, as observed in experiments. Finally, we present evidence for a unique, “wide-open” conformation of Get3. These calculations describe the nucleotide-dependent thermodynamics of Get3 in solution, and improve our understanding of its mechanism in each phase of the GET cycle.
The Molecular Dynamics of Trypanosoma brucei UDP-Galactose-4’-Epimerase: A Drug Target for African Sleeping SicknessChem. Biol. Drug Disc. 80 (2): 173-81 (2012) [PubMed 22487100]
During the past century, several epidemics of human African trypanosomiasis, a deadly disease caused by the protist Trypanosoma brucei, have afflicted sub-Saharan Africa. Over 10 000 new victims are reported each year, with hundreds of thousands more at risk. As current drug treatments are either highly toxic or ineffective, novel trypanocides are urgently needed. The T. brucei galactose synthesis pathway is one potential therapeutic target. Although galactose is essential for T. brucei survival, the parasite lacks the transporters required to intake galactose from the environment. UDP-galactose 4'-epimerase (TbGalE) is responsible for the epimerization of UDP-glucose to UDP-galactose and is therefore of great interest to medicinal chemists. Using molecular dynamics simulations, we investigate the atomistic motions of TbGalE in both the apo and holo states. The sampled conformations and protein dynamics depend not only on the presence of a UDP-sugar ligand, but also on the chirality of the UDP-sugar C4 atom. This dependence provides important insights into TbGalE function and may help guide future computer-aided drug discovery efforts targeting this protein
During the past century, several epidemics of human African trypanosomiasis, a deadly disease caused by the protist Trypanosoma brucei, have afflicted sub-Saharan Africa. Over 10 000 new victims are reported each year, with hundreds of thousands more at risk. As current drug treatments are either highly toxic or ineffective, novel trypanocides are urgently needed. The T. brucei galactose synthesis pathway is one potential therapeutic target. Although galactose is essential for T. brucei survival, the parasite lacks the transporters required to intake galactose from the environment. UDP-galactose 4'-epimerase (TbGalE) is responsible for the epimerization of UDP-glucose to UDP-galactose and is therefore of great interest to medicinal chemists. Using molecular dynamics simulations, we investigate the atomistic motions of TbGalE in both the apo and holo states. The sampled conformations and protein dynamics depend not only on the presence of a UDP-sugar ligand, but also on the chirality of the UDP-sugar C4 atom. This dependence provides important insights into TbGalE function and may help guide future computer-aided drug discovery efforts targeting this protein
Hybrid Finite Element and Brownian Dynamics Method for 1-D Linear and Radial Diffusion-controlled ReactionsJ. Chem. Phys., Vol. 136, Issue 16, 164107 (2012) [PubMed 22559470]
Diffusion is often the rate determining step in many biological processes. Currently, the two main computational methods for studying diffusion are stochastic methods, such as Brownian dynamics, and continuum methods, such as the finite element method. This paper proposes a new hybrid diffusion method that couples the strengths of each of these two methods. The method is derived for a general multidimensional system, and is presented using a basic test case for 1D linear and radially symmetric diffusion systems
Diffusion is often the rate determining step in many biological processes. Currently, the two main computational methods for studying diffusion are stochastic methods, such as Brownian dynamics, and continuum methods, such as the finite element method. This paper proposes a new hybrid diffusion method that couples the strengths of each of these two methods. The method is derived for a general multidimensional system, and is presented using a basic test case for 1D linear and radially symmetric diffusion systems
Multi-timescale conformational dynamics of the SH3 domain of CD2-associated protein using NMR spectroscopy and accelerated molecular dynamicsAngew. Chem. Engl. Ed., Vol. 51, Issue 25, pp. 6103-6106 (2012) [PubMed 22565613]
A complete understanding of the relationship between biological activity and molecular conformation requires an understanding of the thermally accessible potential energy landscape. An extensive set of experimental NMR residual dipolar couplings (RDCs) has been used to determine the conformational behavior of CD2AP SH3C on multiple timescales, using the Gaussian Axial Fluctuation model, and comparison to restraint-free accelerated molecular dynamics simulation. These robust analyses provide a comprehensive description of conformational fluctuations on picosecond to millisecond timescales. While the β-sheets show negligible slow motions, larger amplitude slow dynamics are found in the n-SRC and RT loops that mediate physiological interactions.
A complete understanding of the relationship between biological activity and molecular conformation requires an understanding of the thermally accessible potential energy landscape. An extensive set of experimental NMR residual dipolar couplings (RDCs) has been used to determine the conformational behavior of CD2AP SH3C on multiple timescales, using the Gaussian Axial Fluctuation model, and comparison to restraint-free accelerated molecular dynamics simulation. These robust analyses provide a comprehensive description of conformational fluctuations on picosecond to millisecond timescales. While the β-sheets show negligible slow motions, larger amplitude slow dynamics are found in the n-SRC and RT loops that mediate physiological interactions.
Novel cruzain inhibitors for the treatment of Chagas’s diseaseChem. Biol. Drug Design, Vol. 80, Issue 3, pp. 398-405 (2012) [PubMed 22613098]
The protozoan parasite Trypanosoma cruzi, the etiological agent of Chagas' disease, affects millions of individuals and continues to be an important global health concern. The poor efficacy and unfavorable side effects of current treatments necessitate novel therapeutics. Cruzain, the major cysteine protease of T. cruzi, is one potential novel target. Recent advances in a class of vinyl sulfone inhibitors are encouraging; however, as most potential therapeutics fail in clinical trials and both disease progression and resistance call for combination therapy with several drugs, the identification of additional classes of inhibitory molecules is essential. Using an exhaustive virtual-screening and experimental validation approach, we identify several additional small-molecule cruzain inhibitors. Further optimization of these chemical scaffolds could lead to the development of novel drugs useful in the treatment of Chagas' disease.
The protozoan parasite Trypanosoma cruzi, the etiological agent of Chagas' disease, affects millions of individuals and continues to be an important global health concern. The poor efficacy and unfavorable side effects of current treatments necessitate novel therapeutics. Cruzain, the major cysteine protease of T. cruzi, is one potential novel target. Recent advances in a class of vinyl sulfone inhibitors are encouraging; however, as most potential therapeutics fail in clinical trials and both disease progression and resistance call for combination therapy with several drugs, the identification of additional classes of inhibitory molecules is essential. Using an exhaustive virtual-screening and experimental validation approach, we identify several additional small-molecule cruzain inhibitors. Further optimization of these chemical scaffolds could lead to the development of novel drugs useful in the treatment of Chagas' disease.
LigMerge: A Fast Algorithm to Generate Models of Novel Potential Ligands from Sets of Known BindersChem. Biol. Drug Design, Vol. 80, Issue 3, pp. 358-365 (2012) [PubMed 22594624]
One common practice in drug discovery is to optimize known or suspected ligands in order to improve binding affinity. In performing these optimizations, it is useful to look at as many known inhibitors as possible for guidance. Medicinal chemists often seek to improve potency by altering certain chemical moieties of known/endogenous ligands while retaining those critical for binding. To our knowledge, no automated, ligand-based algorithm exists for systematically "swapping" the chemical moieties of known ligands in order to generate novel ligands with potentially improved potency. To address this need, we have created a novel algorithm called "LigMerge". LigMerge identifies the maximum (largest) common substructure of two three-dimensional ligand models, superimposes these two substructures, and then systematically mixes and matches the distinct fragments attached to the common substructure at each common atom, thereby generating multiple compound models related to the known inhibitors that can be evaluated using computer docking prior to synthesis and experimental testing. To demonstrate the utility of LigMerge, we identify compounds predicted to inhibit peroxisome proliferator-activated receptor gamma, HIV reverse transcriptase, and dihydrofolate reductase with affinities higher than those of known ligands. We are hopeful that LigMerge will be a helpful tool for the drug-design community.
One common practice in drug discovery is to optimize known or suspected ligands in order to improve binding affinity. In performing these optimizations, it is useful to look at as many known inhibitors as possible for guidance. Medicinal chemists often seek to improve potency by altering certain chemical moieties of known/endogenous ligands while retaining those critical for binding. To our knowledge, no automated, ligand-based algorithm exists for systematically "swapping" the chemical moieties of known ligands in order to generate novel ligands with potentially improved potency. To address this need, we have created a novel algorithm called "LigMerge". LigMerge identifies the maximum (largest) common substructure of two three-dimensional ligand models, superimposes these two substructures, and then systematically mixes and matches the distinct fragments attached to the common substructure at each common atom, thereby generating multiple compound models related to the known inhibitors that can be evaluated using computer docking prior to synthesis and experimental testing. To demonstrate the utility of LigMerge, we identify compounds predicted to inhibit peroxisome proliferator-activated receptor gamma, HIV reverse transcriptase, and dihydrofolate reductase with affinities higher than those of known ligands. We are hopeful that LigMerge will be a helpful tool for the drug-design community.
Structure Based Discovery of Novel Druggable Pockets on Rho Family GTPasesPLoS ONE in press (2012)
Latest Advances in Classical Molecular DynamicsIn “Multi-Scale Simulations for Biological Systems,” G. Voth and V. Tozzini (Eds.), Springer (2012)
The dynamic structure of thrombin in solutionBiophys. J., Vol. 103, Issue 1, pp. 79-88 (2012) [PubMed 22828334]
The backbone dynamics of human α-thrombin inhibited at the active site serine were analyzed using R1, R2, and heteronuclear NOE experiments, variable temperature TROSY 2D 1H-15N correlation spectra, and Rex measurements. The N-terminus of the heavy chain, which is formed upon zymogen activation and inserts into the protein core, is highly ordered, as is much of the double beta-barrel core. Some of the surface loops, by contrast, remain very dynamic with order parameters as low as 0.5 indicating significant motions on the ps-ns timescale. Regions of the protein that were thought to be dynamic in the zymogen and to become rigid upon activation, in particular the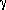 -loop, the 180s
loop, and the Na+ binding site have order parameters below 0.8.
Significant Rex was observed in most of the
-loop, the 180s
loop, and the Na+ binding site have order parameters below 0.8.
Significant Rex was observed in most of the 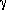 -loop, in
regions proximal to the light chain, and in the β-sheet core.
Accelerated molecular dynamics simulations yielded a molecular ensemble
consistent with measured residual dipolar couplings that revealed
dynamic motions up to milliseconds. Several regions, including the light
chain and two proximal loops, did not appear highly dynamic on the ps-ns
timescale, but had significant motions on slower timescales.
-loop, in
regions proximal to the light chain, and in the β-sheet core.
Accelerated molecular dynamics simulations yielded a molecular ensemble
consistent with measured residual dipolar couplings that revealed
dynamic motions up to milliseconds. Several regions, including the light
chain and two proximal loops, did not appear highly dynamic on the ps-ns
timescale, but had significant motions on slower timescales.
The backbone dynamics of human α-thrombin inhibited at the active site serine were analyzed using R1, R2, and heteronuclear NOE experiments, variable temperature TROSY 2D 1H-15N correlation spectra, and Rex measurements. The N-terminus of the heavy chain, which is formed upon zymogen activation and inserts into the protein core, is highly ordered, as is much of the double beta-barrel core. Some of the surface loops, by contrast, remain very dynamic with order parameters as low as 0.5 indicating significant motions on the ps-ns timescale. Regions of the protein that were thought to be dynamic in the zymogen and to become rigid upon activation, in particular the
iAPBS: A programming interface to Adaptive Poisson-Boltzmann Solver (APBS)Comp. Sci. Discov., Vol. 5, Issue 1 (2012) [PubMed 22905037]
The Adaptive Poisson-Boltzmann Solver (APBS) is a state-of-the-art suite for performing Poisson-Boltzmann electrostatic calculations on biomolecules. The iAPBS package provides a modular programmatic interface to the APBS library of electrostatic calculation routines. The iAPBS interface library can be linked with a FORTRAN or C/C++ program thus making all of the APBS functionality available from within the application. Several application modules for popular molecular dynamics simulation packages - Amber, NAMD and CHARMM are distributed with iAPBS allowing users of these packages to perform implicit solvent electrostatic calculations with APBS.
The Adaptive Poisson-Boltzmann Solver (APBS) is a state-of-the-art suite for performing Poisson-Boltzmann electrostatic calculations on biomolecules. The iAPBS package provides a modular programmatic interface to the APBS library of electrostatic calculation routines. The iAPBS interface library can be linked with a FORTRAN or C/C++ program thus making all of the APBS functionality available from within the application. Several application modules for popular molecular dynamics simulation packages - Amber, NAMD and CHARMM are distributed with iAPBS allowing users of these packages to perform implicit solvent electrostatic calculations with APBS.
Exploring the Photophysical Properties of Molecular Systems using Excited State Accelerated Ab Initio Molecular DynamicsJ. Chem. Theory Comput, Vol. 8, Issue 8, pp. 2752-2761 (2012) [PubMed 22904696]
In the present work, we employ excited state accelerated ab initio molecular dynamics (A-AIMD) to efficiently study the excited state energy landscape and photophysical topology of a variety of molecular systems. In particular, we focus on two important challenges for the modeling of excited electronic states: (i) the identification and characterization of conical intersections and crossing seams, in order to predict different and often competing radiationless decay mechanisms, and (ii) the description of the solvent effect on the absorption and emission spectra of chemical species in solution. In particular, using as examples the Schiff bases formaldimine and salicylidenaniline, we show that A-AIMD can be readily employed to explore the conformational space around crossing seams in molecular systems with very different photochemistry. Using acetone in water as an example, we demonstrate that the enhanced configurational space sampling may be used to accurately and efficiently describe both the prominent features and line-shapes of absorption and emission spectra.
In the present work, we employ excited state accelerated ab initio molecular dynamics (A-AIMD) to efficiently study the excited state energy landscape and photophysical topology of a variety of molecular systems. In particular, we focus on two important challenges for the modeling of excited electronic states: (i) the identification and characterization of conical intersections and crossing seams, in order to predict different and often competing radiationless decay mechanisms, and (ii) the description of the solvent effect on the absorption and emission spectra of chemical species in solution. In particular, using as examples the Schiff bases formaldimine and salicylidenaniline, we show that A-AIMD can be readily employed to explore the conformational space around crossing seams in molecular systems with very different photochemistry. Using acetone in water as an example, we demonstrate that the enhanced configurational space sampling may be used to accurately and efficiently describe both the prominent features and line-shapes of absorption and emission spectra.
Modeling effects of L-type Ca2+ current and Na+-Ca2+ exchanger on Ca2+ trigger flux in rabbit myocytes with realistic t-tubule geometryFront. Comput. Physiol. Med., Vol. 3, Issue 351 (2012) [PubMed 23060801]
The transverse tubular system of rabbit ventricular myocytes consists of cell membrane invaginations (t-tubules) that are essential for efficient cardiac excitation-contraction coupling. In this study, we investigate how t-tubule micro-anatomy, L-type Ca2+ channel (LCC) clustering, and allosteric activation of Na+/Ca2+ exchanger by L-type Ca2+ current affects intracellular Ca2+ dynamics. Our model includes a realistic 3D geometry of a single t-tubule and its surrounding half-sarcomeres for rabbit ventricular myocytes. The effects of spatially distributed membrane ion-transporters (LCC, Na+/Ca2+ exchanger, sarcolemmal Ca2+ pump, and sarcolemmal Ca2+ leak), and stationary and mobile Ca2+ buffers (troponin C, ATP, calmodulin, and Fluo-3) are also considered. We used a coupled reaction-diffusion system to describe the spatio-temporal concentration profiles of free and buffered intracellular Ca2+. We obtained parameters from voltage-clamp protocols of L-type Ca2+ current and line-scan recordings of Ca2+ concentration profiles in rabbit cells, in which the sarcoplasmic reticulum is disabled. Our model results agree with experimental measurements of global Ca2+ transient in myocytes loaded with 50 μM Fluo-3. We found that local Ca2+ concentrations within the cytosol and sub-sarcolemma, as well as the local trigger fluxes of Ca2+ crossing the cell membrane, are sensitive to details of t-tubule micro-structure and membrane Ca2+ flux distribution. The model additionally predicts that local Ca2+ trigger fluxes are at least threefold to eightfold higher than the whole-cell Ca2+ trigger flux. We found also that the activation of allosteric Ca2+-binding sites on the Na+/Ca2+ exchanger could provide a mechanism for regulating global and local Ca2+ trigger fluxes in vivo. Our studies indicate that improved structural and functional models could improve our understanding of the contributions of L-type and Na+/Ca2+ exchanger fluxes to intracellular Ca2+ dynamics.
The transverse tubular system of rabbit ventricular myocytes consists of cell membrane invaginations (t-tubules) that are essential for efficient cardiac excitation-contraction coupling. In this study, we investigate how t-tubule micro-anatomy, L-type Ca2+ channel (LCC) clustering, and allosteric activation of Na+/Ca2+ exchanger by L-type Ca2+ current affects intracellular Ca2+ dynamics. Our model includes a realistic 3D geometry of a single t-tubule and its surrounding half-sarcomeres for rabbit ventricular myocytes. The effects of spatially distributed membrane ion-transporters (LCC, Na+/Ca2+ exchanger, sarcolemmal Ca2+ pump, and sarcolemmal Ca2+ leak), and stationary and mobile Ca2+ buffers (troponin C, ATP, calmodulin, and Fluo-3) are also considered. We used a coupled reaction-diffusion system to describe the spatio-temporal concentration profiles of free and buffered intracellular Ca2+. We obtained parameters from voltage-clamp protocols of L-type Ca2+ current and line-scan recordings of Ca2+ concentration profiles in rabbit cells, in which the sarcoplasmic reticulum is disabled. Our model results agree with experimental measurements of global Ca2+ transient in myocytes loaded with 50 μM Fluo-3. We found that local Ca2+ concentrations within the cytosol and sub-sarcolemma, as well as the local trigger fluxes of Ca2+ crossing the cell membrane, are sensitive to details of t-tubule micro-structure and membrane Ca2+ flux distribution. The model additionally predicts that local Ca2+ trigger fluxes are at least threefold to eightfold higher than the whole-cell Ca2+ trigger flux. We found also that the activation of allosteric Ca2+-binding sites on the Na+/Ca2+ exchanger could provide a mechanism for regulating global and local Ca2+ trigger fluxes in vivo. Our studies indicate that improved structural and functional models could improve our understanding of the contributions of L-type and Na+/Ca2+ exchanger fluxes to intracellular Ca2+ dynamics.
Spectroscopic and Computational Study of Melittin, Cecropin A, and the Hybrid Peptide CM15J. Phys. Chem., Vol. 116, Issue 35, pp. 10600-10608 (2012) [PubMed 22845179]
Antimicrobial peptides (AMPs), such as cecropin A from silk moth, are key components of the innate immune system. They are effective defensive weapons against invading pathogens, yet they do not target host eukaryotic cells. In contrast, peptide toxins, such as honeybee melittin, are nondiscriminating and target both eukaryotic and prokaryotic cells. An AMP-toxin hybrid peptide that is composed of cecropin A and melittin (CM15) improves upon the antimicrobial activity of cecropin A without displaying the nonspecific, hemolytic properties of melittin. Here we report fluorescence and UV resonance Raman spectra of melittin, cecropin A, and CM15 with the goal of elucidating peptide-membrane interactions that help guide specificity. We have probed the potency for membrane disruption, local environment and structure of the single tryptophan residue, backbone conformation near the peptide hinge, and amide backbone structure of the peptides in lipid environments that mimic eukaryotic and prokaryotic membranes. These experimental results suggest that melittin inserts deeply into the bilayer, whereas cecropin A remains localized to the lipid headgroup region. A surprising finding is that CM15 is a potent membrane-disruptor despite its largely unfolded conformation. A molecular dynamics analysis complements these data and demonstrates the ability of CM15 to associate favorably with membranes as an unfolded peptide. This combined experimental–computational study suggests that new models for peptide–membrane interactions should be considered.
Antimicrobial peptides (AMPs), such as cecropin A from silk moth, are key components of the innate immune system. They are effective defensive weapons against invading pathogens, yet they do not target host eukaryotic cells. In contrast, peptide toxins, such as honeybee melittin, are nondiscriminating and target both eukaryotic and prokaryotic cells. An AMP-toxin hybrid peptide that is composed of cecropin A and melittin (CM15) improves upon the antimicrobial activity of cecropin A without displaying the nonspecific, hemolytic properties of melittin. Here we report fluorescence and UV resonance Raman spectra of melittin, cecropin A, and CM15 with the goal of elucidating peptide-membrane interactions that help guide specificity. We have probed the potency for membrane disruption, local environment and structure of the single tryptophan residue, backbone conformation near the peptide hinge, and amide backbone structure of the peptides in lipid environments that mimic eukaryotic and prokaryotic membranes. These experimental results suggest that melittin inserts deeply into the bilayer, whereas cecropin A remains localized to the lipid headgroup region. A surprising finding is that CM15 is a potent membrane-disruptor despite its largely unfolded conformation. A molecular dynamics analysis complements these data and demonstrates the ability of CM15 to associate favorably with membranes as an unfolded peptide. This combined experimental–computational study suggests that new models for peptide–membrane interactions should be considered.
Calcium binding and allosteric signaling mechanisms for sarcoplasmic reticulum Ca2+ ATPaseProtein Science, Vol. 21, Issue 10, pp. 1429-1443 (2012) [PubMed 22821874]
The sarcoplasmic reticulum Ca(2+) ATPase (SERCA) is a membrane-bound pump that utilizes ATP to drive calcium ions from the myocyte cytosol against the higher calcium concentration in the sarcoplasmic reticulum. Conformational transitions associated with Ca(2+) -binding are important to its catalytic function. We have identified collective motions that partition SERCA crystallographic structures into multiple catalytically-distinct states using principal component analysis. Using Brownian dynamics simulations, we demonstrate the important contribution of surface-exposed, polar residues in the diffusional encounter of Ca(2+) . Molecular dynamics simulations indicate the role of Glu309 gating in binding Ca(2+) , as well as subsequent changes in the dynamics of SERCA's cytosolic domains. Together these data provide structural and dynamical insights into a multistep process involving Ca(2+) binding and catalytic transitions.
The sarcoplasmic reticulum Ca(2+) ATPase (SERCA) is a membrane-bound pump that utilizes ATP to drive calcium ions from the myocyte cytosol against the higher calcium concentration in the sarcoplasmic reticulum. Conformational transitions associated with Ca(2+) -binding are important to its catalytic function. We have identified collective motions that partition SERCA crystallographic structures into multiple catalytically-distinct states using principal component analysis. Using Brownian dynamics simulations, we demonstrate the important contribution of surface-exposed, polar residues in the diffusional encounter of Ca(2+) . Molecular dynamics simulations indicate the role of Glu309 gating in binding Ca(2+) , as well as subsequent changes in the dynamics of SERCA's cytosolic domains. Together these data provide structural and dynamical insights into a multistep process involving Ca(2+) binding and catalytic transitions.
Routine access to millisecond timescale events with accelerated molecular dynamicsJ. Chem. Theory Comp., Vol. 8, Issue 9, pp. 2997−3002 (2012) [PubMed 22984356]
In this work, we critically assess the ability of the all-atom enhanced sampling method accelerated molecular dynamics (aMD) to investigate conformational changes in proteins that typically occur on the millisecond time scale. We combine aMD with the inherent power of graphics processor units (GPUs) and apply the implementation to the bovine pancreatic trypsin inhibitor (BPTI). A 500 ns aMD simulation is compared to a previous millisecond unbiased brute force MD simulation carried out on BPTI, showing that the same conformational space is sampled by both approaches. To our knowledge, this represents the first implementation of aMD on GPUs and also the longest aMD simulation of a biomolecule run to date. Our implementation is available to the community in the latest release of the Amber software suite (v12), providing routine access to millisecond events sampled from dynamics simulations using off the shelf hardware.
In this work, we critically assess the ability of the all-atom enhanced sampling method accelerated molecular dynamics (aMD) to investigate conformational changes in proteins that typically occur on the millisecond time scale. We combine aMD with the inherent power of graphics processor units (GPUs) and apply the implementation to the bovine pancreatic trypsin inhibitor (BPTI). A 500 ns aMD simulation is compared to a previous millisecond unbiased brute force MD simulation carried out on BPTI, showing that the same conformational space is sampled by both approaches. To our knowledge, this represents the first implementation of aMD on GPUs and also the longest aMD simulation of a biomolecule run to date. Our implementation is available to the community in the latest release of the Amber software suite (v12), providing routine access to millisecond events sampled from dynamics simulations using off the shelf hardware.
Long timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in Troponin CBiophys. J., Vol. 103, Issue 8, pp. 1784−1789 (2012) [PubMed 23083722]
Troponin (Tn) is an important regulatory protein in the thin-filament complex of cardiomyocytes. Calcium binding to the troponin C (TnC) subunit causes a change in its dynamics that leads to the transient opening of a hydrophobic patch on TnC's surface, to which a helix of another subunit, troponin I (TnI), binds. This process initiates contraction, making it an important target for studies investigating the detailed molecular processes that underlie contraction. Here we use microsecond-timescale Anton molecular dynamics simulations to investigate the dynamics and kinetics of the opening transition of the TnC hydrophobic patch. Free-energy differences for opening are calculated for wild-type Ca(2+)-bound TnC (∼8 kcal/mol), V44Q Ca(2+)-bound TnC (3.2 kcal/mol), E40A Ca(2+)-bound TnC (∼12 kcal/mol), and wild-type apo TnC (∼20 kcal/mol). These results suggest that the mutations have a profound impact on the frequency with which the hydrophobic patch presents to TnI. In addition, these simulations corroborate that cardiac wild-type TnC does not open on timescales relevant to contraction without calcium being bound.
Troponin (Tn) is an important regulatory protein in the thin-filament complex of cardiomyocytes. Calcium binding to the troponin C (TnC) subunit causes a change in its dynamics that leads to the transient opening of a hydrophobic patch on TnC's surface, to which a helix of another subunit, troponin I (TnI), binds. This process initiates contraction, making it an important target for studies investigating the detailed molecular processes that underlie contraction. Here we use microsecond-timescale Anton molecular dynamics simulations to investigate the dynamics and kinetics of the opening transition of the TnC hydrophobic patch. Free-energy differences for opening are calculated for wild-type Ca(2+)-bound TnC (∼8 kcal/mol), V44Q Ca(2+)-bound TnC (3.2 kcal/mol), E40A Ca(2+)-bound TnC (∼12 kcal/mol), and wild-type apo TnC (∼20 kcal/mol). These results suggest that the mutations have a profound impact on the frequency with which the hydrophobic patch presents to TnI. In addition, these simulations corroborate that cardiac wild-type TnC does not open on timescales relevant to contraction without calcium being bound.
Comment on “Molecular driving forces of the pocket-ligand hydrophobic association” by G. Graziano, Chem. Phys. Lett. 533 (2012) 95Chem. Phys. Lett., Vol. 555, Issue ?, pp. 306−309 (2012)
We comment on a study by G. Graziano recently published in Chem. Phys. Lett. 533 (2012) 95, which presented an alternative interpretation of our previous study on hydrophobic cavity-ligand association (J. Chem. Theory Comput. 6 (2010) 2866). Here, we show why this interpretation is not appropriate in the context of cavity-ligand binding. We also demonstrate that a thorough understanding of an association process can be achieved only after considering all entropic and enthalpic contributions, including also those that cancel with each other.
We comment on a study by G. Graziano recently published in Chem. Phys. Lett. 533 (2012) 95, which presented an alternative interpretation of our previous study on hydrophobic cavity-ligand association (J. Chem. Theory Comput. 6 (2010) 2866). Here, we show why this interpretation is not appropriate in the context of cavity-ligand binding. We also demonstrate that a thorough understanding of an association process can be achieved only after considering all entropic and enthalpic contributions, including also those that cancel with each other.
Dynamics of Plasmodium falciparum Enoyl-ACP Reductase and Implications on Drug DiscoveryProtein Sci., Vol. 21, Issue 11, pp. 1734−1745 (2012) [PubMed 22969045]
Enoyl-acyl carrier protein reductase (ENR) is a crucial enzyme in the type II fatty acid synthesis pathway of many pathogens such as Plasmodium falciparum, the etiological agent of the most severe form of malaria. Because of its essential function of fatty acid double bond reduction and the absence of a human homologue, PfENR is an interesting drug target. Although extensive knowledge of the protein structure has been gathered over the last decade, comparatively little remains known about the dynamics of this crucial enzyme. Here, we perform extensive molecular dynamics simulations of tetrameric PfENR in different states of cofactor and ligand binding, and with a variety of different ligands bound. A pocket-volume analysis is also performed, and virtual screening is used to identify potential druggable hotspots. The implications of the results for future drug-discovery projects are discussed.
Enoyl-acyl carrier protein reductase (ENR) is a crucial enzyme in the type II fatty acid synthesis pathway of many pathogens such as Plasmodium falciparum, the etiological agent of the most severe form of malaria. Because of its essential function of fatty acid double bond reduction and the absence of a human homologue, PfENR is an interesting drug target. Although extensive knowledge of the protein structure has been gathered over the last decade, comparatively little remains known about the dynamics of this crucial enzyme. Here, we perform extensive molecular dynamics simulations of tetrameric PfENR in different states of cofactor and ligand binding, and with a variety of different ligands bound. A pocket-volume analysis is also performed, and virtual screening is used to identify potential druggable hotspots. The implications of the results for future drug-discovery projects are discussed.
The Binding Mechanism, Multiple Binding Modes, and Allosteric Regulation of Staphylococcus aureus SrtA Probed by Molecular Dynamics SimulationsProtein Sci., Vol. 21, Issue 12, pp. 1858−1871 (2012) [PubMed 23023444]
Sortase enzymes are vitally important for the virulence of gram-positive bacteria as they play a key role in the attachment of surface proteins to the cell wall. These enzymes recognize a specific sorting sequence in proteins destined to be displayed on the surface of the bacteria and catalyze the transpeptidation reaction that links it to a cell wall precursor molecule. Because of their role in establishing pathogenicity, and in light of the recent rise of antibiotic-resistant bacterial strains, sortase enzymes are novel drug targets. Here, we present a study of the prototypical sortase protein Staphylococcus aureus Sortase A (SrtA). Both conventional and accelerated molecular dynamics simulations of S. aureus SrtA in its apo state and when bound to an LPATG sorting signal (SS) were performed. Results support a binding mechanism that may be characterized as conformational selection followed by induced fit. Additionally, the SS was found to adopt multiple metastable states, thus resolving discrepancies between binding conformations in previously reported experimental structures. Finally, correlation analysis reveals that the SS actively affects allosteric pathways throughout the protein that connect the first and the second substrate binding sites, which are proposed to be located on opposing faces of the protein. Overall, these calculations shed new light on the role of dynamics in the binding mechanism and function of sortase enzymes.
Sortase enzymes are vitally important for the virulence of gram-positive bacteria as they play a key role in the attachment of surface proteins to the cell wall. These enzymes recognize a specific sorting sequence in proteins destined to be displayed on the surface of the bacteria and catalyze the transpeptidation reaction that links it to a cell wall precursor molecule. Because of their role in establishing pathogenicity, and in light of the recent rise of antibiotic-resistant bacterial strains, sortase enzymes are novel drug targets. Here, we present a study of the prototypical sortase protein Staphylococcus aureus Sortase A (SrtA). Both conventional and accelerated molecular dynamics simulations of S. aureus SrtA in its apo state and when bound to an LPATG sorting signal (SS) were performed. Results support a binding mechanism that may be characterized as conformational selection followed by induced fit. Additionally, the SS was found to adopt multiple metastable states, thus resolving discrepancies between binding conformations in previously reported experimental structures. Finally, correlation analysis reveals that the SS actively affects allosteric pathways throughout the protein that connect the first and the second substrate binding sites, which are proposed to be located on opposing faces of the protein. Overall, these calculations shed new light on the role of dynamics in the binding mechanism and function of sortase enzymes.
Finite Element Estimation of Protein-Ligand Association Rates with Post-Encounter Effects: Applications to Calcium binding in Troponin C and SERCAComp. Sci. Disc., 5 014015 [PubMed 23293662]
We introduce a computational pipeline and suite of software tools for the approximation of diffusion-limited binding based on a recently developed theoretical framework. Our approach handles molecular geometries generated from high-resolution structural data and can account for active sites buried within the protein or behind gating mechanisms. Using tools from the FEniCS library and the APBS solver, we implement a numerical code for our method and study two Ca(2+)-binding proteins: Troponin C and the Sarcoplasmic Reticulum Ca(2+) ATPase (SERCA). We find that a combination of diffusional encounter and internal 'buried channel' descriptions provide superior descriptions of association rates, improving estimates by orders of magnitude.
We introduce a computational pipeline and suite of software tools for the approximation of diffusion-limited binding based on a recently developed theoretical framework. Our approach handles molecular geometries generated from high-resolution structural data and can account for active sites buried within the protein or behind gating mechanisms. Using tools from the FEniCS library and the APBS solver, we implement a numerical code for our method and study two Ca(2+)-binding proteins: Troponin C and the Sarcoplasmic Reticulum Ca(2+) ATPase (SERCA). We find that a combination of diffusional encounter and internal 'buried channel' descriptions provide superior descriptions of association rates, improving estimates by orders of magnitude.
Molecular Basis of Calcium-sensitizing Mutations of the Human Cardiac Troponin C Regulatory Domain: A multi-scale simulation studyPLoS Comp. Biol., Vol. 8, Issue 11, e1002777 (2012) [PubMed 23209387]
Troponin C (TnC) is implicated in the initiation of myocyte contraction via binding of cytosolic Ca2+ and subsequent recognition of the Troponin I switch peptide. Mutations of the cardiac TnC N-terminal regulatory domain have been shown to alter both calcium binding and myofilament force generation. We have performed molecular dynamics simulations of engineered TnC variants that increase or decrease Ca2+ sensitivity, in order to understand the structural basis of their impact on TnC function. We will use the distinction for mutants that are associated with increased affinity and for those mutants with reduced Ca2+ affinity. Our studies demonstrate that for GOF mutants V44Q and L48Q, the structure of the physiologically-active site II Ca2+ binding site in the Ca2+-free (apo) state closely resembled the Ca2+-bound (holo) state. In contrast, site II is very labile for LOF mutants E40A and V79Q in the apo form and bears little resemblance with the holo conformation. We hypothesize that these phenomena contribute to the increased association rate, kon , for the GOF mutants relative to LOF. Furthermore, we observe significant positive and negative positional correlations between helices in the GOF holo mutants that are not found in the LOF mutants. We anticipate these correlations may contribute either directly to Ca2+ affinity or indirectly through TnI association. Our observations based on the structure and dynamics of mutant TnC provide rationale for binding trends observed in GOF and LOF mutants and will guide the development of inotropic drugs that target TnC.
Troponin C (TnC) is implicated in the initiation of myocyte contraction via binding of cytosolic Ca2+ and subsequent recognition of the Troponin I switch peptide. Mutations of the cardiac TnC N-terminal regulatory domain have been shown to alter both calcium binding and myofilament force generation. We have performed molecular dynamics simulations of engineered TnC variants that increase or decrease Ca2+ sensitivity, in order to understand the structural basis of their impact on TnC function. We will use the distinction for mutants that are associated with increased affinity and for those mutants with reduced Ca2+ affinity. Our studies demonstrate that for GOF mutants V44Q and L48Q, the structure of the physiologically-active site II Ca2+ binding site in the Ca2+-free (apo) state closely resembled the Ca2+-bound (holo) state. In contrast, site II is very labile for LOF mutants E40A and V79Q in the apo form and bears little resemblance with the holo conformation. We hypothesize that these phenomena contribute to the increased association rate, kon , for the GOF mutants relative to LOF. Furthermore, we observe significant positive and negative positional correlations between helices in the GOF holo mutants that are not found in the LOF mutants. We anticipate these correlations may contribute either directly to Ca2+ affinity or indirectly through TnI association. Our observations based on the structure and dynamics of mutant TnC provide rationale for binding trends observed in GOF and LOF mutants and will guide the development of inotropic drugs that target TnC.
Allosteric Networks in Thrombin Distinguish Procoagulant vs. Anticoagulant ActivitiesProc. Natl. Acad. Sci. USA, Vol. 109, Issue 52, pp. 21216−21222 (2012) [PubMed 23197839]
The serine protease alpha-thrombin is a dual-action protein that mediates the blood-clotting cascade. Thrombin alone is a procoagulant, cleaving fibrinogen to make the fibrin clot, but the thrombin-thrombomodulin (TM) complex initiates the anticoagulant pathway by cleaving protein C. A TM fragment consisting of only the fith and sixth EGF-like domains (TM56) is sufficient to bind thrombin, but the presence of the fourth EGF-like domain (TM456) is critical to induce the anticoagulant activity of thrombin. Crystallography of the thrombin-TM456 complex revealed no significant structural changes in the thrombin, suggesting that TM4 may only provide a scaffold for optimal alignment of protein C for its cleavage by thrombin. However, a variety of experimental data have suggested that the presence of TM4 may affect the dynamic properties of the active site loops. In the present work, we have used both conventional and accelerated molecular dynamics simulation to study the structural dynamic properties of thrombin, thrombin:TM56 and thrombin:TM456 across a broad range of time scales. Two distinct, yet interrelated allosteric pathways are identified that mediate both the pro- and anticoagulant activities of thrombin. One allosteric pathway, which is present in both thrombin:TM56 and thrombin:TM456, directly links the TM5 domain to the thrombin active site. The other allosteric pathway, which is only present on slow time scales in the presence of the TM4 domain, involves a complex extended network of correlated motions linking the TM4 and TM5 domains and the active site loops of thrombin.
The serine protease alpha-thrombin is a dual-action protein that mediates the blood-clotting cascade. Thrombin alone is a procoagulant, cleaving fibrinogen to make the fibrin clot, but the thrombin-thrombomodulin (TM) complex initiates the anticoagulant pathway by cleaving protein C. A TM fragment consisting of only the fith and sixth EGF-like domains (TM56) is sufficient to bind thrombin, but the presence of the fourth EGF-like domain (TM456) is critical to induce the anticoagulant activity of thrombin. Crystallography of the thrombin-TM456 complex revealed no significant structural changes in the thrombin, suggesting that TM4 may only provide a scaffold for optimal alignment of protein C for its cleavage by thrombin. However, a variety of experimental data have suggested that the presence of TM4 may affect the dynamic properties of the active site loops. In the present work, we have used both conventional and accelerated molecular dynamics simulation to study the structural dynamic properties of thrombin, thrombin:TM56 and thrombin:TM456 across a broad range of time scales. Two distinct, yet interrelated allosteric pathways are identified that mediate both the pro- and anticoagulant activities of thrombin. One allosteric pathway, which is present in both thrombin:TM56 and thrombin:TM456, directly links the TM5 domain to the thrombin active site. The other allosteric pathway, which is only present on slow time scales in the presence of the TM4 domain, involves a complex extended network of correlated motions linking the TM4 and TM5 domains and the active site loops of thrombin.
